发病时间:不清楚
容易跟小儿糖原贮积病Ⅰ型混淆的疾病有哪些?小儿糖原贮积病Ⅰ型混淆的原因是什么?
补充说明:容易跟小儿糖原贮积病Ⅰ型混淆的疾病有哪些?小儿糖原贮积病Ⅰ型混淆的原因是什么?
a******7 2011-05-29 10:38
小儿糖原贮积病Ⅰ型 黏多糖贮积症Ⅱ型 皮肤 综合征 骨骼畸形
我要咨询
相关问题
医生回答(1)

王朝 主治医师 三甲
提问
容易跟小儿糖原贮积病Ⅰ型混淆的疾病如下:
1.黏多糖贮积症Ⅱ型
病因是艾杜糖醛酸-2-硫酸酯酶缺乏。临床上有重型(A)和轻型(B)。由于酶缺乏使硫酸皮肤素(DS)和硫酸类肝素降解障碍,在体内储留并由尿中排出,二者的排出量比为1:1。
较为少见。其中A型的病情较重。患者全部为男性,多于2~6岁起病。临床表现与Hurler综合征相似,但出现时间较晚,进展较缓慢。B型患者病情较轻,有的听力和角膜可均正常,亦无骨骼畸形。
2.黏多糖贮积症Ⅲ型
酶的缺乏各亚型不同。ⅢA型为硫酸酰胺酶(旧名称类肝素-N-硫酸酯酶)缺乏,ⅢB为α-N-乙酰葡糖胺酶缺乏,ⅢC为N-乙酰基转移酶缺乏,ⅢD为葡糖胺-6-硫酸酯酶缺乏
极为少见。虽然本型可有4种不同的酶缺乏,但其临床表现非常相似,主要为进行性的智力减退,其中以黏多糖贮积症ⅢA型的临床进展较快。
3.黏多糖病Ⅳ型(Morquio氏病),有两个亚型。其病因为ⅣA为半乳糖-6-硫酸酯酶缺乏,ⅣB为β-D半乳糖酶缺乏。
面容及智力正常。学步较晚,行走时步态蹒跚不稳。短颈、耸肩。出牙时间较晚,牙列不整齐,牙齿缺乏光泽。角膜混浊可早在儿童期开始出现。听力呈进行性损害。
4.黏多糖贮积症Ⅴ型
现认为该型即为黏多糖贮积症Ⅰ型的Seheie型,与Hrular综合征不同之处表现为无严重的角膜混浊,且混浊为周边性,患者智力正常,身材正常或稍矮,寿命基本正常,但有多毛,关节强直。背柱、头颅X线示仅有轻微改变。
5.黏多糖贮积症Ⅵ型
黏多糖病Ⅵ型又称Maroteaux-Lamy综合征。为N-乙酰半乳糖胺-4-硫酸酯酶缺乏,临床上分重型和轻型。
极为罕见。临床表现与黏多糖贮积症Ⅰ型相似,但患者的智力正常。一般从2~3岁开始出现生长迟缓。颅骨缝闭合较早,可出现脑积水,并引起颅高压症状和痉挛性偏瘫。角膜混浊出现较早,有进行性听力损害,严重者有失明和耳聋。
6.黏多糖贮积症Ⅶ型
黏多糖病Ⅶ型是β-D-葡糖醛酸酶缺乏,为常染色体隐性遗传,该酶基因位于7q21.2-q22区。
极罕见。特殊面容在出生后不久即开始逐渐出现。一般智力正常,角膜混浊及听力损害较常见。多有肝脾肿大,通常不累及心脏,无腹外疝。上肢较短,骨骼发育不良,可有鸡胸、膝外翻等骨骼畸形。
7.黏多糖病Ⅷ型
黏多糖病Ⅷ型1978年开始报道,病因是由于N-乙酰氨基葡糖-6-硫酸酯酶缺乏,
临床表现有黏多糖病Ⅲ型和Ⅳ型的共同特征,有侏儒,智能落后,脏器受累和骨骼畸形,无角膜混浊。
2011-05-29 10:46
举报向医生提问
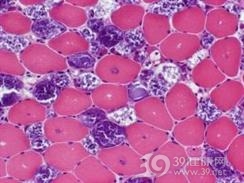
糖原贮积病Ⅰ型又称VonGeirk病、葡萄糖-6-磷酸酶缺陷症、粘多糖症Ⅰ型、脂质黏多糖病Ⅰ型。黏多糖贮积症患者由于过多的黏多糖贮积于骨、软骨等组织或器官内,从而影响到这些组织或器官的正常发育,多余的黏多糖从尿中排出。 1968最早由Spmnger描述本症,本病多见于婴幼儿,年长儿也可见到。

复方苁蓉益智胶囊
益智养肝,活血化浊,健脑增智。用于轻、中度血管性痴呆肝肾亏虚兼痰瘀阻络证。症见智力减退、思维迟钝、神情呆滞、健忘,或喜怒不定、腰膝酸软、头晕耳鸣、失眠多梦等。

小儿感冒颗粒
疏风解表,清热解毒。用于小儿风热感冒,症见发热、头胀痛、咳嗽痰黏、咽喉肿痛;流感见上述证候者。
盐酸纳洛酮舌下片
用于急性酒精中毒,限于步态不稳定、话多、不连贯、欣快、共济失调、感知迟钝、困倦、嗜睡,但不伴有昏迷及生命体征改变的急性酒精中毒的酩酊状态。 本品主要成份及其化学名称为:17-烯丙基-4,5;环氧基-3,14-羟基吗啡喃-6-酮盐酸盐二水合物。基结构式为:分子式:C19H21NO4;HCl;2H2O分子量:399.87。

净石灵胶囊
补肾,利尿,排石。用于治疗肾结石、输尿管结石、膀胱结石以及由结石引起的肾盂积水、尿路感染等。







