发病时间:不清楚
请问小儿黏多糖贮积症症状有哪些?
补充说明:请问小儿黏多糖贮积症症状有哪些?
a******7 2011-06-01 09:41
我要咨询
相关问题
医生回答(1)

李瑾 主治医师 三甲
提问
小儿黏多糖贮积症有多种症状,收集了该病的所有症状,希望对您有帮助:
黏多糖是一种复合大分子,主要由糖醛酸和己糖胺构成,分布在结缔组织的基质内,为软骨、角膜、血管壁和皮下组织的重要成分。因此黏多糖代谢异常累及全身器官,黏多糖贮积症发病后,软骨肌膜、肌腱、血管、心脏瓣膜肌肉、脑膜、网状内皮组织及皮下组织等胶原组织的成纤维细胞均肿胀,其内充以黏多糖颗粒肝、脾、肾、淋巴结和某些内分泌器官的实质细胞内,亦有类似物质沉积。中枢神经和周围神经之神经节细胞亦肿胀,但充盈的物质主要为神经苷脂,而黏多糖含量很少或不含有黏多糖。
患者一般出生时正常,随年龄增大,临床症状逐渐明显,由于各型病情轻重不一,又有各自的临床特征,在诊断中需鉴别。其共同特征是:
A)在出生1年左右出现生长落后。除Ⅳ型和Ⅵ型外,患者都伴有智能落后。
B)身材矮小和特殊面容,表情淡漠,头大、面部丑陋,眼裂小,眼距宽,鼻梁低平,鼻孔大,唇厚,前额和双颧突出,毛发多而发际低,颈短,大部分有角膜混浊。关节进行性畸变,胸廓畸形,脊柱后凸或侧凸,膝外翻、爪形手。
C)早期出现肝、脾肿大,耳聋,心脏增大等。
D)由于黏多糖在血管壁的沉积,可突发冠状动脉闭塞或心肌梗死。
黏多糖病是常染色体遗传病。由于组织细胞内溶酶体酶(如1-艾杜糖苷酸酶、硫酸脂酶、N-乙酰-D-氨基葡糖苷酶、α-N-乙酰转移酶β-半乳糖苷酶等)缺陷造成黏多糖降解不全,并在溶酶体内堆聚,尿不完全代谢产物排出增加导致软骨、结缔组织、心脏、中枢神经等功能障碍。由于缺陷酶的不同,引起细胞内贮存的黏多糖粘脂质也不均一,目前对引起黏多糖病的酶缺陷都已鉴定,共分为6型。
1.黏多糖病Ⅰ型:包括ⅠS和ⅠH两型。本病是常染色体隐性遗传病,α-L-艾杜糖醛酸苷酶基因已被鉴定,位于染色体4p16.3,有14个外显子,并在其中发现了不少基因突变。临床表型与基因型分析发现,α-L-艾杜糖醛酸苷酶基因突变导致酶活性严重缺乏者称为黏多糖病Ⅰ-H型,如在编码区70,或402出现终止密码。基因突变导致酶活性中度或轻度下降者,在临床上分类为黏多糖病Ⅰ-S型。
黏多糖病Ⅰ-H型(Hurler综合征) 是较严重的一种类型,常在10岁左右死亡,病因为缺乏α-L-艾杜糖醛酸苷酶,导致硫酸皮肤素和硫酸肝素在体内积聚,全身脏器如角膜、软骨、骨骼、皮肤、心肌内膜、血管结缔组织等均受累。临床有智能低下,面容丑陋(巨头宽大鼻缘、饱满的颊、厚唇、大舌头、粗毛及多毛症、驼背、关节活动受限、爪样手等),肝脾肿大,骨骼病变,心血管病变,角膜混浊和耳聋。末梢血白细胞、淋巴细胞可见到异染的大小不等,形态不同的深染颗粒,有时呈空泡状,尿排出大量酸性黏多糖(>100mg/d,正常为3~25mg/d)。正常人尿中黏多糖排出量为每天5~15mg,其中硫酸皮肤素和硫酸乙酰肝素各约占10%,硫酸角质素仅占少量(0.1mg/kg)。
黏多糖病Ⅰ-S型(Scheie综合征),原先分类为黏多糖病Ⅴ型,属中等度严重类型黏多糖病,遗传类型和致病基因同黏多糖病Ⅰ-H型。智能发育正常,临床症状一般在5岁后出现。
2.黏多糖病Ⅱ型(Hunter综合征) 临床重型与黏多糖Ⅰ-H型相似,在2~6岁起病,有特殊面容和骨骼畸形,但脊椎无鸟嘴样畸形。无角膜混浊。患者智能落后,呈进行性耳聋,可发生充血性心力衰竭,肝脾肿大。
本型为X连锁隐性遗传,病因是艾杜糖醛酸硫酸酯酶缺陷,使硫酸皮肤素和硫酸肝素代谢障碍。致病基因已克隆,位于染色体Xq28区,在脆性X综合征区域附近,有9个外显子,基因分析发现较多病人有大片段缺失,其他形式有点突变、小片段缺失或插入。临床表型与基因型较为符合。有严重病变,如在核苷酸1129位上插入22个碱基或有基因缺失者病情较重,基因有点突变者相对属轻型临床表现。
3.黏多糖病Ⅲ型(Sanfilippo综合征) 临床可分为4种亚型,分别由4种不同的酶缺陷所引起。ⅢA型为硫酸酰胺酶缺乏,ⅢB型为α-N-乙酰己糖胺酶缺乏,ⅢC型为α-葡糖胺N-乙酰转移酶缺乏,ⅢD型为N-乙酰葡糖胺-6-硫酸酯酶缺乏。上述4种酶都是硫酸肝素降解所需要的酶,因此,当这些酶缺乏时均引起硫酸肝素在体内积聚,同时尿中排出量增加。临床上患儿在1岁内发育尚正常,以后逐渐出现语言、行为障碍,生长发育落后,在儿童期神经系统退行性病变较明显,有肝脾肿大,疝气,面容丑陋,关节强直等。面部及骨骼畸形较轻,但运动与精神异常发生更早更严重。
本病4型均为常染色体隐性遗传性疾病,导致硫酸肝素在体内积聚。ⅢD型的致病基因葡糖胺-6-硫酸酯酶已克隆,位于染色体12q14。
4.黏多糖病Ⅳ型(Morquio综合征) 临床特征与黏多糖病Ⅰ-H型相似,但无智能障碍。有明显的生长障碍明显侏儒样,骨骼畸形,X线呈典型的黏多糖病表现,脊椎有鸟嘴样突出改变,椎骨扁平,飘带肋骨,鸡胸等,面容丑陋,鼻矮、口大、牙齿发育不良,角膜混浊。青春发育可正常,随年龄增长出现脊髓压迫症状,晚期出现压迫性截瘫和呼吸麻痹。没有内脏黏多糖贮存表现。
黏多糖病Ⅳ型有2种亚型,黏多糖病Ⅳ型A为N-乙酰半乳糖胺-6-硫酸酯酶缺乏,黏多糖病Ⅳ型B为β-半乳糖酶缺乏,使硫酸角质素和硫酸软骨素降解障碍,导致这些物质在细胞与组织中积聚。两亚型的表型相同,都属常染色体隐性遗传性疾病。N-乙酰半乳糖胺-6-硫酸酯酶的全长cDNA已克隆,基因定位于染色体16q24.3,并在此基因上发现了一些突变位点。β-半乳糖酶基因也已克隆,定位于染色体3q21.33,并找到了突变位点。
5.黏多糖病Ⅴ型(Maroteaux-Lamy综合征) 临床表现同黏多糖病Ⅰ-H型相似,面部畸形轻骨骼病变轻,智力发育正常。尿中排出大量硫酸皮肤素,致病基因为N-乙酰半乳糖胺-4-硫酸酯酶,基因定位于染色体5q13-5q14,属常染色体隐性遗传性疾病。
6.黏多糖病Ⅵ型 临床表现同黏多糖病Ⅰ-H型,但个体轻重程度不一,变异较大,轻者可无智能落后。本型为常染色体隐性遗传性疾病,因β-葡萄糖醛酸酶缺乏,导致4/6硫酸软骨素在体内沉积。基因定位于染色体7q21.11,有12个外显子。 A)罕见,有明显的以上各种表现型与骨畸形。根据临床特征、特殊面容和体征、X线片表现以及尿黏多糖阳性,可以做出诊断。B)家族史 有黏多糖病人的家族史对早期诊断有帮助。
2011-06-01 09:57
举报向医生提问
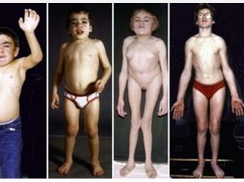
黏多糖病(mucopolysaccharidosis,MPS)是一组先天性遗传病,因黏多糖降解酶缺乏使酸性黏多糖不能完全降解,导致黏多糖积聚在机体不同组织,造成骨骼畸形、智能障碍等一系列临床症状和体征。根据不同酶不同引起的不同症状,可分为6型。