发病时间:不清楚
先天脊肌萎缩症
补充说明:先天脊肌萎缩症
a******W 2015-12-28 16:00
我要咨询
精选回答(1)

赵英帅 主治医师 河南省人民医院 三甲
擅长:擅长报告解读、幽门螺旋杆菌感染、高血压、高血脂、高尿酸、冠心病、糖尿病、胃肠炎、便秘、感冒、肺结节、甲状腺结节等疾病的综合诊疗
提问
主要是预防或治疗SMA的各种并发症,预防肺部感染及褥疮、营养不良、骨骼畸形、行动障碍和精神社会性问题,如伴有呼吸功能不全需用人工呼吸器,保证气道通畅,改善呼吸功能。长期卧床可造成坠积;误吸也可造成肺炎,预防肺炎的有效措施有辅助咳嗽、胸部叩击治疗及间歇正压通气,即使在没有急性呼吸道感染的情况下,患者也需保持良好的肺部通气状态,预防发生进行性肺不张。一旦有效肺活量(FVC)下降,即使肢体或躯干的肌力无明显改变,发生肺炎的危险性也会增高。
2015-12-28 15:59
举报相关问题
医生回答(4)

李志恒 主治医师 三甲
提问
先天脊肌萎缩症一般是指脊髓性肌萎缩症,是一类由脊髓前角运动神经元变性导致肌无力、肌萎缩的疾病,属于常染色体隐性遗传病。
脊髓性肌萎缩症的发病原因尚不明确,可能是因为遗传因素、环境因素等原因导致的,患者会出现肢体无力、肌肉萎缩、肌肉纤颤、腱反射减弱等症状。患者需要及时到医院进行检查,然后在医生的指导下使用诺西那生钠注射液、利司扑兰口服溶液等药物进行治疗。
患者在治疗期间要注意休息,保持充足的睡眠时间,避免熬夜。同时,患者还要注意保持良好的心态,积极配合医生进行治疗,有利于病情的恢复。饮食上可以适当进食富含蛋白质、维生素等营养物质的食物,如鸡蛋、芹菜等,有利于补充身体所需营养,促进病情恢复。
2015-12-28 15:59
举报程银兵 主治医师 三甲
擅长:全科
提问
又称SMA-Ⅲ型,也称为Kugelberg-Welander病Wohlfart-Kugelberg-Welander综合征或轻度SMA。是SMA中表现最轻的一类。本病在儿童晚期或青春期出现症状,开始为步态异常下肢近端肌肉无力。缓慢进展。渐累及下肢远端和上双肢。可存活至成人期。表现为神经元性近端肌萎缩。能行走的SMA-Ⅲ型患儿可出现蹒跚步态,腰椎前突,腹部凸起,腱反射可有可无。维持独立行走的时间与肌无力的发病年龄密切相关2岁前发病者将在15岁左右不能行走,2岁后发病者可一直保持行走能力至50岁左右。
2015-12-28 15:59
举报段英达 主治医师 三甲
擅长:全科
提问
病因尚未明确。根据家系分析大多数学者认为是常染色体隐性遗传,小部分为基因突变引起,是否有生化的缺陷尚不清楚。1.婴儿型脊髓性肌萎缩也称为SMA-I型或Werdnig-Hoffmann病。本型为3型中最为严重,部分病例在宫内发病,胎动变弱,半数在出生时或出生后的最初几个月即可发病,且几乎均在5个月内发病。罕见能存活1年,这些患儿在胎儿期已有症状,胎动减少,出生后即有明显四肢无力,喂养困难及呼吸困难。2.中间型脊髓性肌萎缩也称为SMA-Ⅱ型、中间型SMA或慢性SMA,发病较Ⅰ型稍迟,多于1岁内起病,进展缓慢。患儿在6-8个月时生长发育正常多数病例表现以近端为主的严重肌无力,下肢重于上肢;许多Ⅱ型患儿可独坐,少数甚至可以在别人的帮助下站立或行走,但不能独自行走;多发性微小肌阵挛是主要表现;呼吸肌吞咽肌不受累,面肌不受累括约肌功能正常。本型具有相对良性的病程。生存期超过4年可存活至青春期以后。
2015-12-28 15:59
举报李焕晨 主治医师 三甲
擅长:全科
提问
肌肉萎缩指骨骼肌体积的缩小,可由于肌纤维变细或消失,是许多神经肌肉疾病的重要症状和体征.对于肌萎缩患者,首先应注意有无废用性因素(如骨折石膏固定后,关节病或其他疾病长期卧床影响肢体活动等原因引起),应积极诊断,认真治疗. 表现为局限性一侧颞肌,嚼肌萎缩,张口时下颌偏向病侧,可同时伴有面部感觉及角膜反射减退或消失 表现为额或面颊局部的斑块性萎缩,皮肤色素较深,掐之皮下组织紧张,神经系统检查无异常 青春期起病,缓慢进行性面肌萎缩伴闭眼不紧,鼓腮或吹口哨不能,肱部肌肉萎缩,上肢举手时肩胛骨呈翼样突起,无感觉异常
2015-12-28 15:59
举报向医生提问
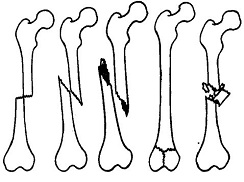
所谓骨折,顾名思义,就是指骨头或骨头的结构完全或部分断裂。多见于儿童及老年人,中青年也时有发生。病人常为—个部位骨折,少数为多发性骨折。经及时恰当处理,多数病人能恢复原来的功能,少数病人可留有不同程度的后遗症。骨折发生后,离医院较近者,可直接送医院或叫救护车。离医院比较远的病人,必须进行简单的处理,以防在送医院途中加重病情,甚至造成不可逆的后果。